
不知道学习什么新知识的时候,可以温习旧的知识。
这篇文章是对原核生物基因组一般分析思路的梳理和回顾。需要强调的是,文章内容只针对单个基因组的分析,不包含基因组重测序的结果分析,比较基因组的分析内容也大都不包含在其中。
大多数原核生物的基因组比较简单,通常是一个环状的染色体结构,以及若干的质粒组成(非必须)。单个基因组测序研究的主要目的,是为了得到目标物种的基因组描述信息,以及对基因组上各种基因发挥功能的探索。
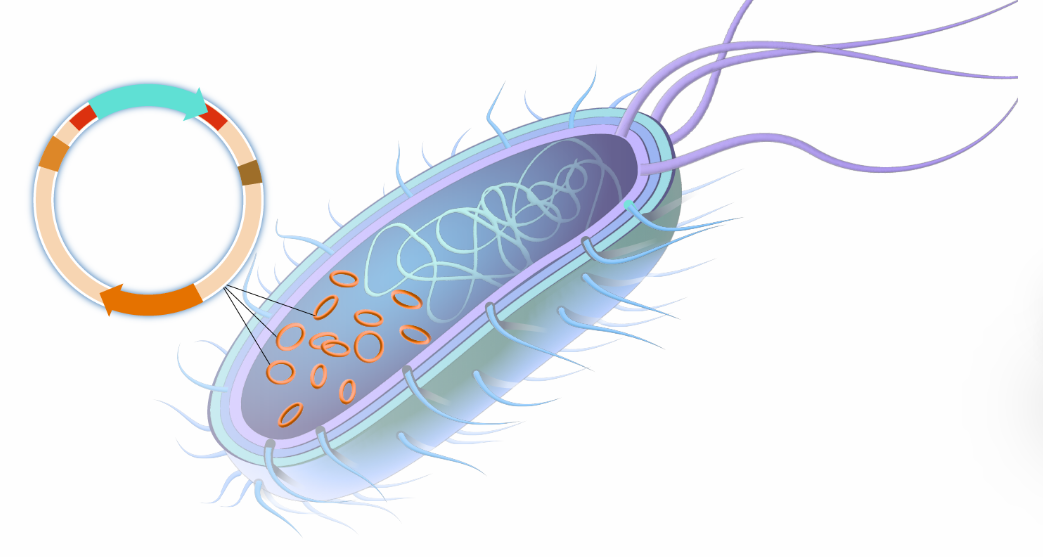
针对我写在这里的研究目的而言,在原始测序数据下机到基因组功能注释的整个过程中,重要的分析步骤包含了如下这些:
原始测序数据质控 –> 基因组组装 –> 组装结果评估 –> 基因预测 - 元件分析 –> 基因功能注释
在对每个步骤梳理之前,关于原核生物的基因组有一些必要的背景知识需要提前说明:
基因组的完整度决定了后续分析的表现,而能否获得完整的基因组取决于测序数据的类型以及组装过程的质量。
NCBI 的基因组数据库 Genome,对原核基因组的组装水平可以大致的划分为 contig、scaffold、chromosome、complete 这 4 类【Genome - NCBI - NLM (nih.gov)】。一定程度上,contig ≈ scaffold,chromosome ≈ complete。chromosome 或者 complete 水平的基因组,暂且称为完整图。相对应的 contig 或者 scaffold 水平的基因组,暂且称为框架图。那么如何理解这几个水平基因组的关系呢,尝试从以下 3 点来看:
- 从低到高: contig 是组装的起点,scaffold 是 contig 的延伸,chromosome 是 scaffold 的进一步整合,而 complete 则代表了基因组组装的最高水平。
- 从局部到整体: contig 反映局部序列信息,scaffold 反映更大范围的序列结构,chromosome 则反映整个基因组的整体结构。
- 从不完整到完整: contig 和 scaffold 之间存在间隙,表示序列信息不完整,而 chromosome 和 complete 则表示基因组序列是完整的,是环状的染色体结构,可能也包含一些质粒。
| 下面这张图(来自:[Filling in the gaps: GapPredict can complement repertoire of tools used to resolve missing DNA sequences in genome assemblies | Genome Sciences Centre (bcgsc.ca)](https://www.bcgsc.ca/news/filling-gaps-gappredict-can-complement-repertoire-tools-used-resolve-missing-dna-sequences)),可以部分,但是直观的反映上述信息: |
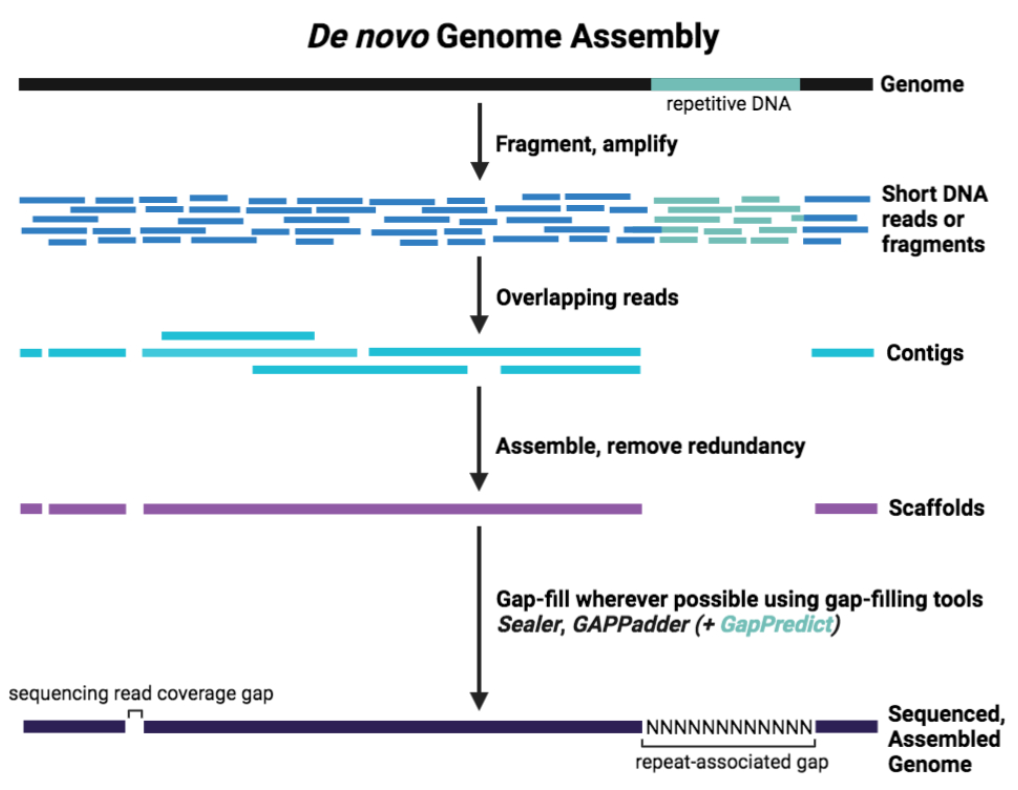
- 需要粗暴的一个理解内容是:
只有二代短读长测序数据的话,只可以得到 contig 或者 scaffold 水平的基因组框架图。
二代 + 三代(长读长测序数据,例如 pacbio 或者 nanopore)的测序数据,可以得到 complete 水平的基因组完整图。
接下来回到我们这次分享的主题,再次重申,本次基因组分析重要的步骤包含如下这些:
原始测序数据质控 –> 基因组组装 –> 组装结果评估 –> 基因预测 - 元件分析 –> 基因功能注释
原始测序数据质控
在上面提到了基因组的完整度和测序数据类型有关,具体包含了短读长双端测序数据,长读长测序数据的代表 pacbio 和 nanopore 的数据,那么针对不同的测序数据,质控方式也是不一样的。
- 短读长测序数据质控
通常,测序的原始数据通常会包含的问题有接头污染,碱基质量低和短序列等问题,这里我们利用 fastp 做序列质控的工作。简单原理讲的话,fastp 可以使用内置的接头序列来识别接头污染的序列;利用滑动窗口的方式来计算序列的平均质量值,并将平均质量值低于阈值的序列过滤掉;同时可以利用阈值设定来过滤短的序列。
输入文件是双端 fq 原始测序数据,对应输出是质控后的序列数据。运行方式如下,多数参数按照默认即可:
fastp -i sample_1.fastq.gz \
-o sample_clean.1.fastq.gz \
-I sample_2.fastq.gz \
-O sample_clean.2.fastq.gz \
-w 8 -h sample.html -j sample.json
- 长读长测序数据质控
Pacbio 和 Nanopore 最终收获的测序数据都是单条长读长序列,不论是 pacbio 的荧光化学信号还是 nanopore 的电流信号,收获的读长并不像二代短读长长度一致,以及三代测序方法获得的长读长序列,错误率要比二代短读长高。因此 fastp 质控的方法并不适用于三代测序数据。
这里我们选用 chopper(参考:wdecoster/chopper (github.com))对长读长序列进行质控,该工具适用于 PacBio 或 ONT 等长读长测序数据,可过滤和修剪 fastq 文件。作为新手入门,应该说是很好的工具可以来利用:
chopper -q 10 -l 500 -i reads.fastq > filtered_reads.fastq
基因组组装
我之前对几乎所有的原核生物基因组组装工具做了一些测试,并对工具做了相关的总结,你可以从这里看到:wanjinhu/Microbial-genome: all about microbial genome (github.com)。从组装效果、效率以及软件帮助文档的详细程度,不论是只有二代测序数据,还是二代 + 三代测序数据,我更加推荐 Unicycler(rrwick/Unicycler: hybrid assembly pipeline for bacterial genomes (github.com))。
同时,如果你想要更好的了解原核基因组组装的过程,我强烈推荐 Unicycler 的说明帮助文档,非常值得一读。其作者 Ryan Wick 的工作专注于细菌基因组的组装,并开发了一系列非常好用的细菌基因组相关的工具,值得大家一试。

Unicycler 的结果提供了从原始 reads 到组装结果过程中的有用文件,其运行也非常方便,一般你可以调整保留最终组装 contig 的最短长度,cpu 等运行参数。
只有短读长双端测序数据:
unicycler -1 short_reads_1.fastq.gz -2 short_reads_2.fastq.gz -o output_dir
只有长读长单链数据:
unicycler -l long_reads.fastq.gz -o output_dir
二代+三代测序数据:
unicycler -1 short_reads_1.fastq.gz -2 short_reads_2.fastq.gz -l long_reads.fastq.gz -o output_dir
我推崇 Unicycler 的一个点在于,对完整基因组组装后,会对基因组的起始位点做矫正,默认 dnaA 或者 repA 基因为起始位点。当然,如果利用了其他的组装软件,最终的组装结果我推荐一款工具来做起始位点矫正,以及完整环状基因组判定的矫正,即:gbouras13/dnaapler: Reorients assembled microbial sequences (github.com)。
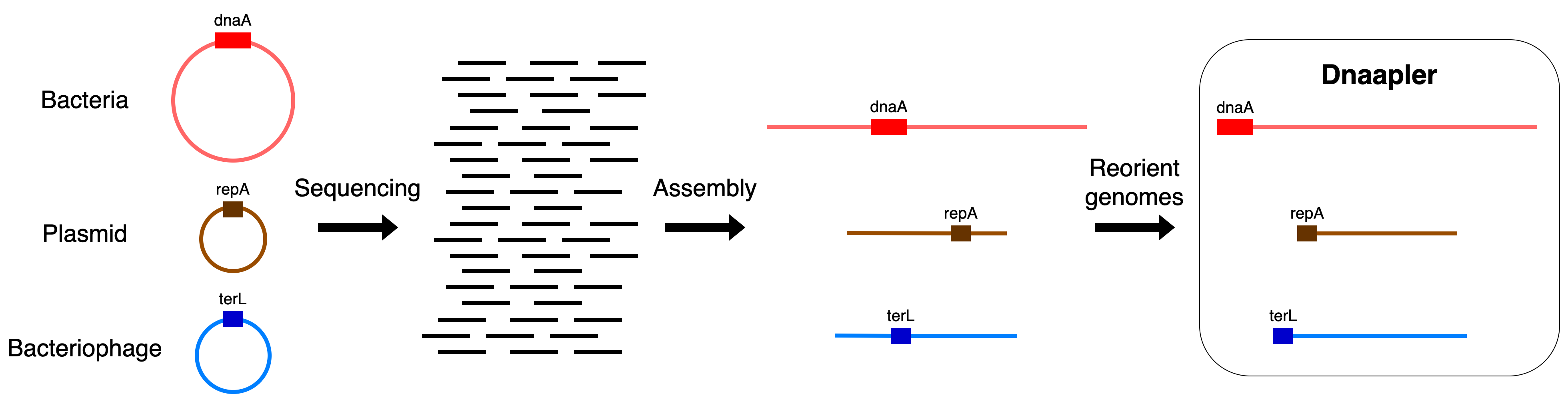
# 对混合基因组等
dnaapler all -i input_mixed_contigs.fasta -o output_directory_path -p my_bacteria_name -t 8
# 对完整染色体基因组
dnaapler chromosome -i input_chromosome.fasta -o output_directory_path -p my_bacteria_name -t 8
组装结果评估
基因组组装完成后,需要对基因组做一个完整的评估。可以从以下内容来评估:
- 基因组完整度和污染程度评估
这里推荐 checkM,是一款用于评估微生物基因组质量的常用工具,原理是通过比较基因组中单拷贝基因的存在、缺失以及拷贝数来估计基因组的完整度和污染程度。
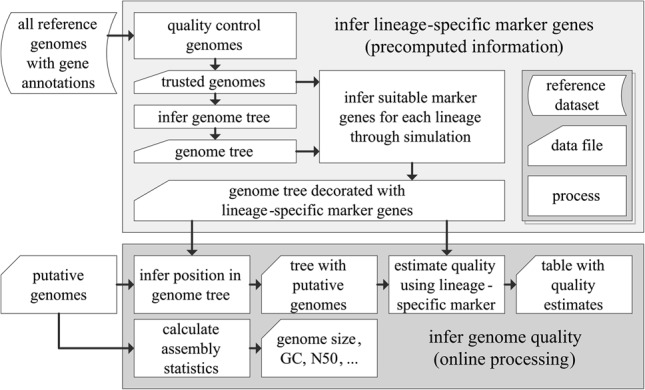
checkM 工作流程,可以参考:CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes - PMC (nih.gov)。
单拷贝基因在物种的基因组中通常只出现一次,如果基因组中绝大多数的单拷贝基因都存在,那么这个基因组就认为是比较完整的,如果缺失了大量的单拷贝基因,则说明该基因组可能是不完整的。checkM 用指标完整性(Completeness)来表示,即基因组中包含的单拷贝基因占所有单拷贝基因的比例。
对于污染程度的评估,如果一个基因组中出现了多个拷贝的单拷贝基因,或者出现了不属于该物种的单拷贝基因,就说明了该基因组可能受到了污染。checkM 用指标污染度(Contamination)来表示,即基因组中包含的非预期基因的比例。
完整性(Completeness)和污染度(Contamination)的数值结果可以通过下表来理解并做常规判断:

checkM 的标准运行方式如下,利用子程序 lineage_wf 进行分析:
# 两个位置参数分别是包含基因组的文件<bin folder>及以及结果输出文件夹<output folder>,-x参数控制你的基因组后缀是什么,比如fna/fasta/fa等
checkm lineage_wf -x fna <bin folder> <output folder>
# 具体示例
checkm lineage_wf -t 32 -x fasta ./ ./checkm
输出结果在 checkm/storage/bin_stats_ext.tsv,示例的分析结果显示,Completeness: 100.0, Contamination: 0.3456221198156682。可以看到完整度为 100%,表示在 checkM 的单拷贝基因参考数据库中,该基因组可以全部被标记到;污染度为 0.35%,表示不属于该基因组物种的基因比例只有不到 0.5%,污染几乎没有。在这一点来看,该基因组的完整度和污染度评估不错。
- 基因组 GC 含量变化评估
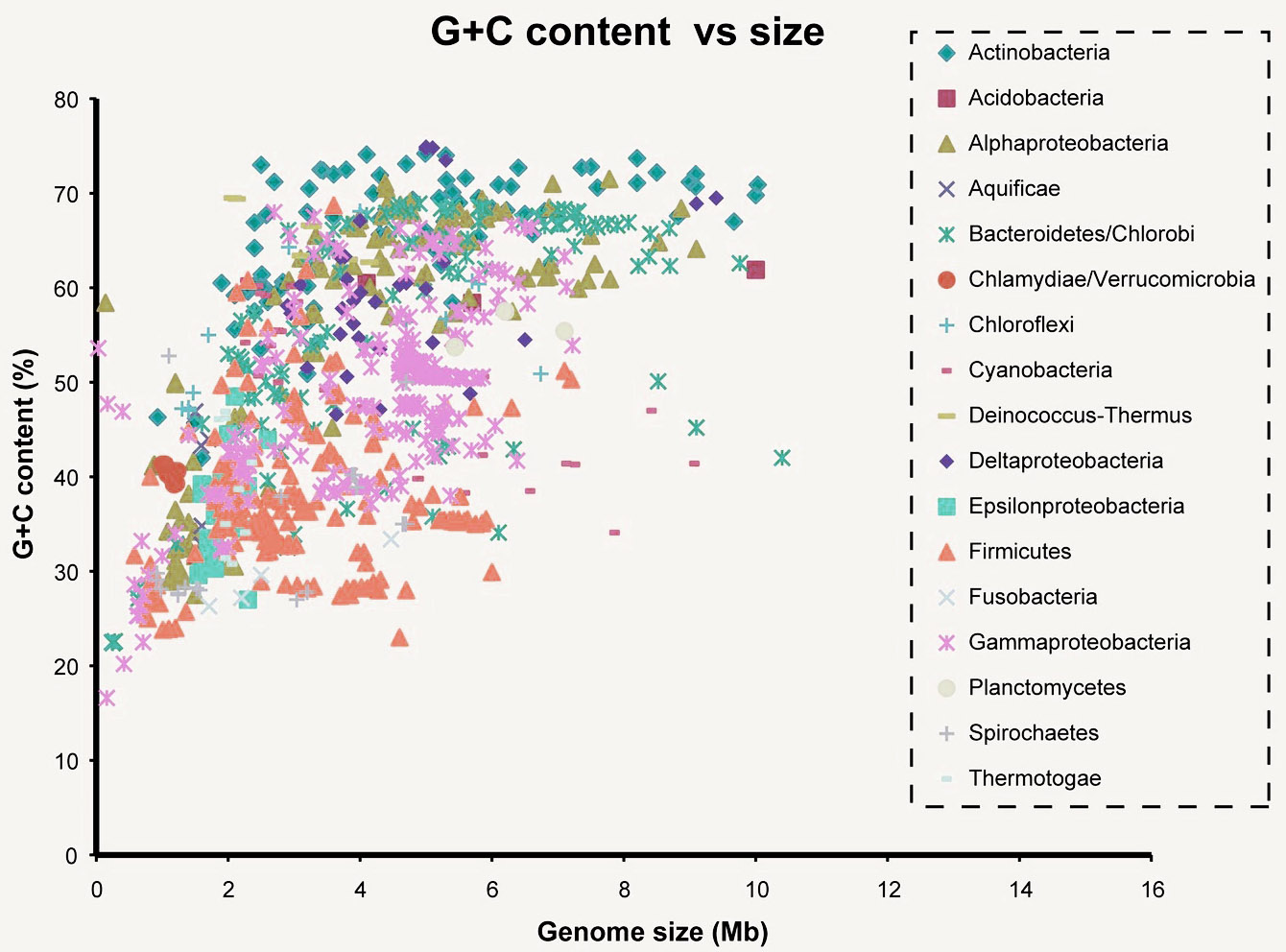
在物种进化过程中,基因组 GC 含量是一个重要的特征。来自同一祖先的细菌,其 GC 含量往往较为接近,这是因为它们共享了一部分共同的基因组特征。相对应的,亲缘关系较远的物种,GC 含量往往也不一样。因此在同一环境下分离得到的菌株,如果基因组不同区域的 GC 含量有较大的差异,也就意味着可能受到了污染。
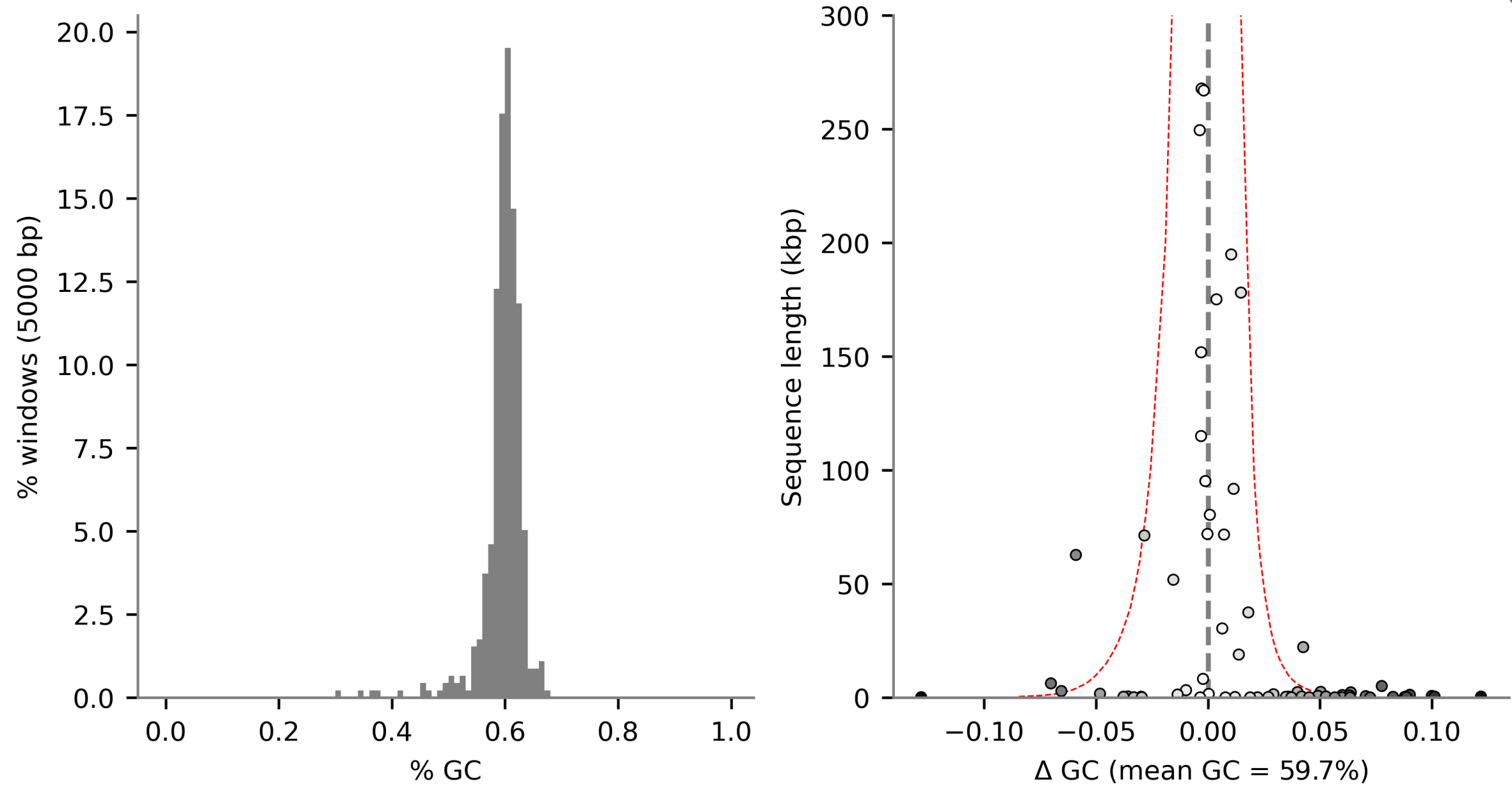
利用固定长度的滑动窗口对一个基因组进行 GC 含量变化的分析,可以对基因组是否污染进行检测。同样 checkM,利用子程序 gc_plot,可以做这项工作。需要注意,该分析是上一个要点中 checkM 运行的下游分析。
# 两个位置参数分别是基因组文件夹<bin folder>和输出文件夹<plot fold>,95为统计偏差95%水平
checkm gc_plot <bin folder> <plot fold> 95
# 具体示例
checkm gc_plot ./ ./checkm/plots 95
示例输出结果比如下图,左图是 5000bp 滑动窗口,统计 GC 平均含量分布的直方图,可以看到是单峰的结果,表明 GC 含量一致,如果是出现多峰,则表明该基因组有污染。右图表示滑动窗口不同位置的 GC 含量与整个基因组 GC 含量的偏差,红色虚线表示与平均 GC 的预期偏差,可以看到偏差的分布和左图也是一致的单峰状态。那么这里的 GC 含量评估结果显示该基因组不存在污染。
- N50/N90值评估
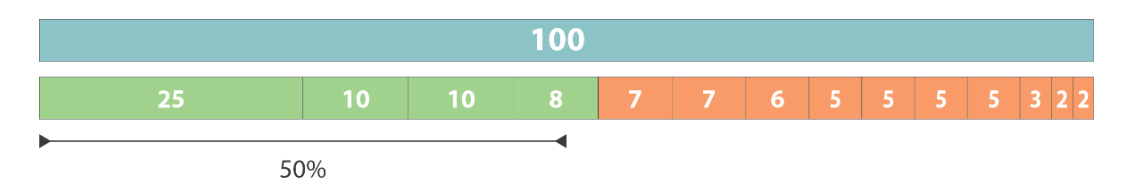
该评估点主要针对非完整的框架图基因组的长度和连续性,比如 contig 或者 scaffold 水平的基因组。针对框架图水平的基因组来说,基因组碎片化越少,丢失的信息也就越少。
我们将组装得到的所有的 contig 或 scaffold 按照长度从大到小排序,当累加长度达到总长度的一半时,最后一个被累加进去的 contig 或 scaffold 的长度就是 N50。如上图所示的话,基因组假如是 100,contig 排序是 (25, 10, 10, 8 , 7, 7 , 6 , 5, 5, 5, 5, 3, 2, 2 ) = 100,总长度达到 50% 时候的 contig 的长度是 8,也就意味着 N50 为 8。
在一些文章中你可能会看到 L50 这个指标,L50 指的是达到 50% 长度时,前面这些 contig 的长度之和。上面这张图的 L50 = 25+10+10+8 = 53。N90 和 L90 也是同样的计算原理。
那么 N50 和 N90 的值如何来评估框架图质量的好坏呢?通过上面的计算过程,显而易见的一个结论是,当组装结果碎片化越小,其 N50 的值越大,说明组装得到的 contig 或 scaffold 越长,组装的连续性越好,组装质量越高。也就是说,N50 反映了基因组中较长连续序列的比例。那么两个组装软件对同一个基因组进行组装时,应该选择 N50/N90 值较大的结果。
基因预测 - 元件分析
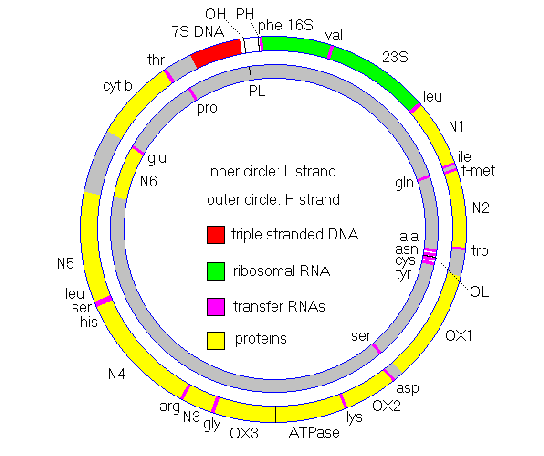
我这里标题的描述并不准确,实际的意思是对整个基因组上的组件信息做预测,比如关注基因组上编码基因、rRNA、tRNA 等的分布情况。同时,对一些特殊的元件也可以做相应的预测分析,比如有没有 sRNA、重复序列、抗菌肽,以及前噬菌体等可移动遗传元件等等。这些都是根据实际的研究目的来决策分析的。
其中对于编码基因,rRNA,tRNA 等常规元件的分析,用 prokka 分析很方便,prokka 其实也是封装的其他软件包来对不同元件进行分析:
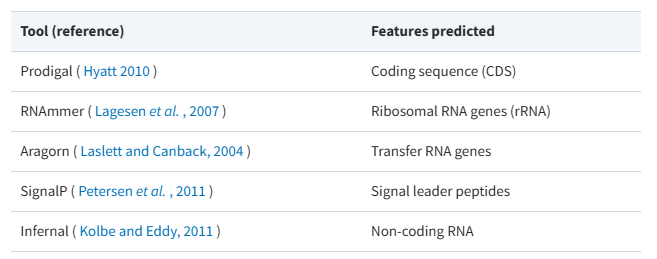
# 输入组装好的基因组,选择物种为bacteria,选择用于 DNA 翻译的遗传密码<gcode>,locustag为可选参数
prokka --outdir result --force --prefix prokka --kingdom bacteria --gcode 11 --cpus 16 /input_dir/assembly.fasta --locustag PLKJGNKB
prokka 输出结果包含了常见的基因组格式的文件,可以在下游分析中使用到,格式和 NCBI 中提供的是一致的:
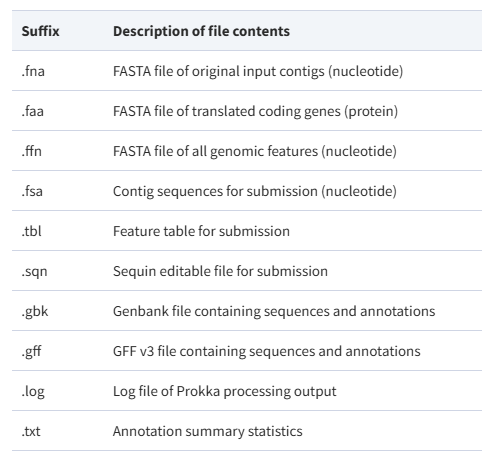
对于其他元件的分析,比如 sRNA、重复序列、抗菌肽和前噬菌体等等,由于分析方式种类很多,我在这里就不一一个介绍了。有机会的话,我会再梳理写一篇文章来说明这个问题。
基因功能注释
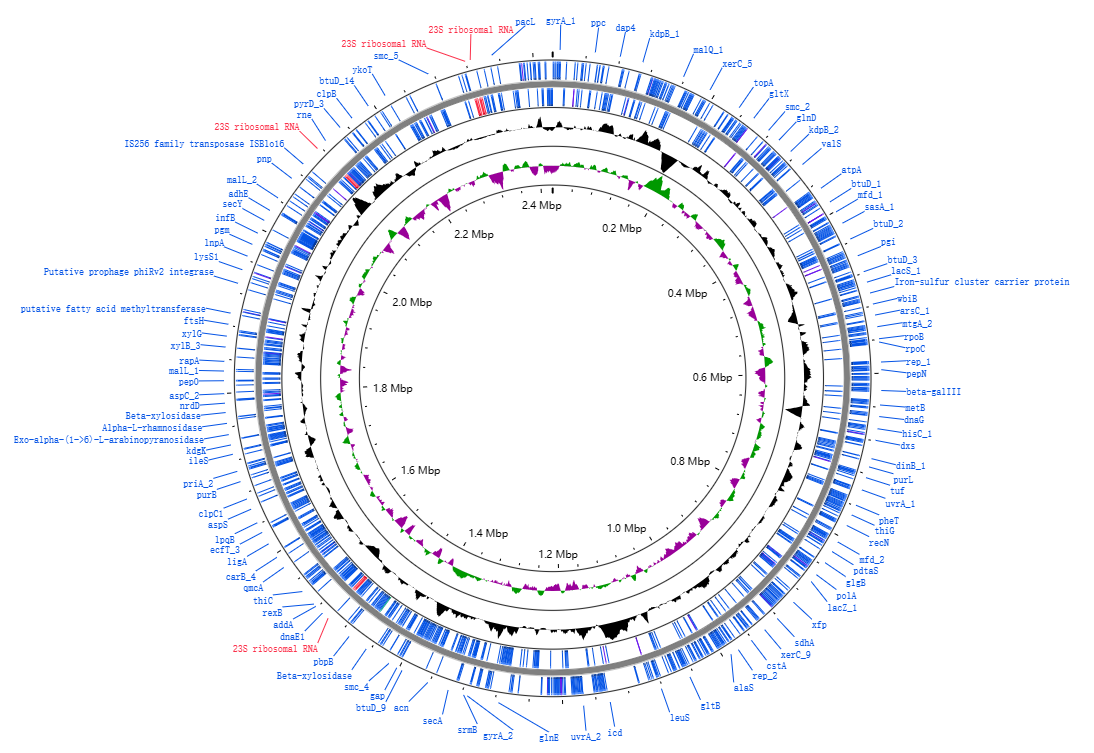
对于 tRNA、rRNA 的信息,上一步 prokka 运行的时候,分别利用 Aragorn 和 RNAmmer 进行了预测。而这个模块的内容主要指获取基因组上编码基因(CDS)的功能注释信息。
很显然的是,prokka 最终的结果是包含了蛋白功能的注释结果,在 gff/gbk 等注释文件中,对于可注释到的基因有明确的对应信息,而注释不到的蛋白,则用假定蛋白(hypothetical protein)作为标识。
prokka 对编码蛋白的注释,采用了完整的策略,主要是利用了 UniProt 数据库的蛋白注释信息,利用 BLAST 以及隐马尔可夫模型的方式去检索,对于符合阈值参数的蛋白给予完整的注释信息,而匹配不到的蛋白则给予假定蛋白的标识。

分析到这里的话,你完全可以利用 prokka 的注释结果做一些描述性结果了,那为什么研究要利用很多功能数据库去做比对注释呢?
对于同一个蛋白而言,不同的数据库注释到的信息应该是一致的。问题在于,如果不是“博览群书”,现在给你一个蛋白,你很难完全把该蛋白的功能描述清楚,更不用说一个基因组上至少有上千的基因信息。同时,prokka 注释到的假定蛋白,我们也很想知道其潜在的功能是什么。
众多的功能数据库,就是把这些蛋白按照其发挥的功能做了分门别类的整理:
功能性专注的数据库,例如:碳水化合物活性酶数据库 CAZY,是专门关注于合成或者分解复杂碳水化合物和糖复合物酶类的数据库;CARD 是一个综合抗生素耐药性数据库,提供与抗菌素耐药性分子基础相关的数据、模型和算法等信息;VFDB 则是专注于细菌病原体的毒力因子信息的数据库。像是类似的功能数据库还有很多,往往是专注于某个具体的功能,根据你自己的研究目的来选择性的使用其中的一些。
功能更加全面或者宽泛的数据库,例如:NR 非冗余蛋白数据库(NCBI 提供);Swiss-Prot 非冗余基因数据库(EBI 提供);COG 直系同源蛋白分类数据库;GO 同源基因分类数据库;以及鼎鼎有名的京都基因和基因组百科全书 KEGG 数据库,提供了更为全面的基因组及基因信息,包括了代谢通路、同源基因、酶活、化合物、化学反应关系等更全面的研究数据库。
很显然,这么多数据库不需要你都做一遍注释分析,不仅浪费了很大的资源,还有很多冗余的信息。总之就是要根据自己的研究目的来挑选适合的数据库来做分析。
而具体做分析的过程,就是拿你在上一步预测到的蛋白序列去和这些数据库中的信息做比对,从而知道基因的相关功能。一些可以利用在线分析的方式进行,如果要线下分析,就要对数据库做整理,而数据库的整理是很关键的,这可能又是另一个文章故事了。
通过上述内容的梳理,希望你能掌握原核生物基因组分析的一般分析思路。即:
原始测序数据质控 –> 基因组组装 –> 组装结果评估 –> 基因预测 - 元件分析 –> 基因功能注释
这篇文章的不足之处
只能作为原核生物基因组一般分析思路与内容的理解,指导做具体的分析内容只是部分可行,一些内容没写具体的代码;
现在纯粹只做单个基因组分析其实很少见了,基本都会涉及到比较基因组的内容,比如研究菌株进化关系、基因水平转移事件等等,而这些内容在这篇文章中没有体现;
在<基因预测 - 元件分析>和<基因功能注释>这两个模块,我其实写的很泛泛,是因为可选的内容实在是太多,不知道从哪里入手,我感觉整个文章是比较虎头蛇尾吧。
总之,在我自己总结梳理的时候,很多内容还是回顾了一下,对已有知识的巩固有很大帮助。
如果这篇文章对你也有一定的帮助作用,那简直太棒太酷了!
文档信息
- 本文作者:Wanjin Hu (胡万金)
- 本文链接:https://wanjinhu.github.io/2024/10/21/article-bacterial-genome/
- 版权声明:自由转载-非商用-非衍生-保持署名(创意共享3.0许可证)