
这篇文章回答一个问题:初学者如何理解并学会宏基因组数据的初步分析过程?
广义上的宏基因组学,如果我们理解为宏组学的话,具体研究的不同是体现在微生物群落遗传物质组成的不同阶段,比如宏基因组学(包括扩增子),宏转录组,蛋白组和代谢组等等。狭义上的宏基因组学呢,指的就是某个样本中所有微生物的 DNA 序列数据了,是该领域目前最为成熟的分支。
上述提到的这些宏组学方法,关键字是“宏”。比如说在常规的肠道微生物研究中,选取了人体宿主在某个状态下的粪便样本,我们可以利用宏基因组学来获取这些粪便样本中所有微生物的信息;对应地,如果是利用基因组学,则需要先从粪便样本中根据自己的实验条件,分离出某种微生物,再具体对这个微生物(单一菌株培养到指数期的状态)的基因组进行分析研究。可以看到,基因组的研究对象是某个物种,宏基因组的研究对象则是某个样本。
通过基因组,可以得到某个物种上所有 DNA 序列数据,进一步分析获取该物种的基因功能;宏基因组则是得到某个样本中所有微生物的 DNA 数据,再将这些 DNA 序列数据转化为微生物物种组成以及基因功能组成的信息。
所以宏基因组学的初步研究目的,就是获取样本中的所有微生物信息。后续的其他分析都是基于此,我们举一个简单的研究示例来说明:
肥胖和正常人群由于其本身基因特点、饮食习惯和生活方式等的差异,可能会导致其肠道微生物的种类以及这些微生物所发挥的功能有所差异,进一步导致了这两组人群可能患有胃肠道疾病的风险不同。这可以作为我们的研究假设。
为了验证假设,我们收集了肥胖和健康组两组人群的粪便样本,并进行了宏基因组测序,通过各种分析手段得到了两组人群的微生物物种组成和功能组成数据,再通过差异检验分析结果显示:A、B、C 三个微生物物种,以及 a、b、c 这三个功能在肥胖和健康人群中具有显著差异性,而这些差异物种和功能又和胃肠道疾病具有显著的相关性。那么至少在相关性结果上,我们发现了这两组人群肠道微生物物种和功能的差异特点。
其他研究也是类似的,可以是临床微生物、环境微生物、农业微生物等等,利用宏基因组学都是类似的研究思路。当然实际的研究科学问题中肯定比我刚才举的例子要复杂的多,涉及到的研究方法也会更多。但是如果是利用了宏基因组学技术的话,各个样本中的物种、功能组成数据结果就是初步分析的重点。
less is more,目前宏基因组数据分析的流程、方法、软件等很多,也有很多一站式、封装好的的流程模式。关于组学数据分析流程,我提倡的学习思维应该是模块化的,每一个分析模块都可以是独立的,也就意味着可以替换成不同的方法和软件,这容易让你思考每一个步骤在做什么,更有助于理解。
关于软件的使用,宏基因组数据的分析使用命令行方式来运行,确定好程序的输入文件、输出文件、选择合适的运行参数即可。作为初学者,先不要纠结具体分析参数(除了数据库等之外的)改成什么比较合适,按照默认参数运行一些项目后,就会有相应的经验。
那我在这里的讲解只是作抛砖引玉,利用的是PE150双端测序数据。整个分析过程的输入数据是原始测序fq数据,输出结果是样本物种及功能的组成表。有如下分析内容概览:
- 原始fq数据质控
- 宿主序列信息去除
- 微生物物种组成分析
- 序列组装
- 基因预测
- 去除冗余基因并生成非冗余基因集
- 功能注释
- 微生物功能组成分析
- 功能结果整理
需要明确的是,上述这些分析内容不是顺序执行,可以按照下面的简易流程图来理解:
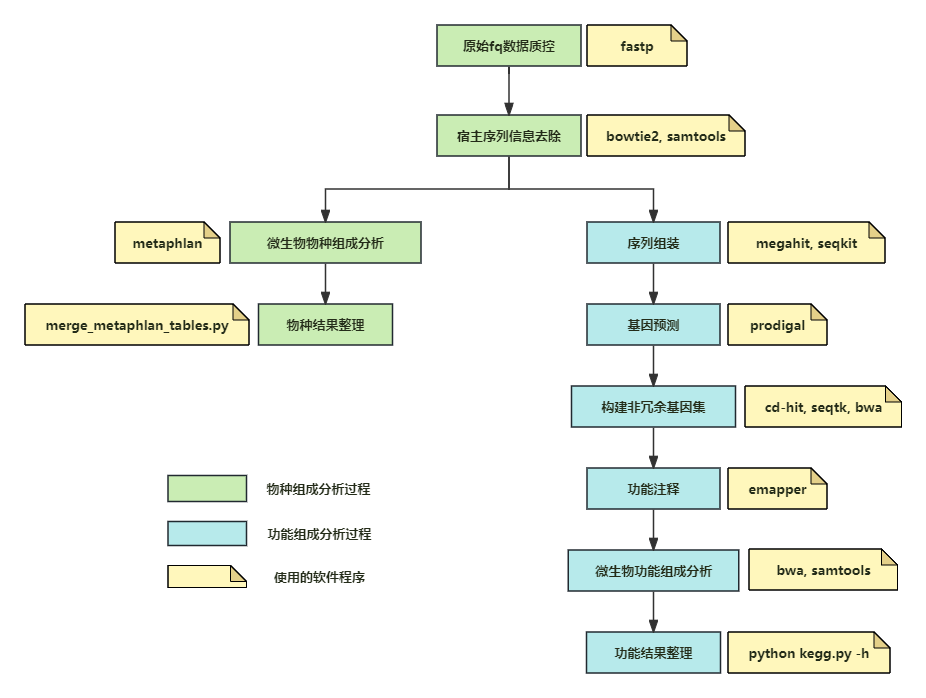
步骤1:原始fq数据质控
测序的原始数据通常会包含的问题有接头污染,碱基质量低和短序列等问题,这里我们利用 fastp 做序列质控的工作。简单原理讲的话,fastp 可以使用内置的接头序列来识别接头污染的序列;利用滑动窗口的方式来计算序列的平均质量值,并将平均质量值低于阈值的序列过滤掉;同时可以利用阈值设定来过滤短的序列。
输入文件是双端fq原始测序数据,对应输出是质控后的序列数据。如下:
fastp -i sample_1.fastq.gz \
-o sample_clean.1.fastq.gz \
-I sample_2.fastq.gz \
-O sample_clean.2.fastq.gz \
-w 8 -h sample.html -j sample.json
步骤2:宿主序列信息去除
宏基因组研究的目标是微生物群落中所有微生物的基因组信息,而不希望被其他信息所干扰。但是在实际采集样本过程中,往往会存在宿主序列的污染,比如采集的粪便样本中会有人体肠内壁的脱落物基因信息等。而宿主序列的污染会影响后续的数据分析,比如导致微生物组成的统计分析结果不准确,进一步影响差异检验的分析结果。
这里基于宿主基因组比对的方法,用bowtie2将上一步得到的质控后的序列与宿主基因组比对,得到比对的sam文件。再利用samtools过滤掉比对上的序列,那么得到的双端序列就是不包含宿主基因信息的高质量序列。如下:
参考基因组的fa序列从NCBI的Genome数据库进行下载,比如人的参考基因组可以下载最新的GRCh38.p14版本:Homo sapiens - NCBI - NLM (nih.gov)。
# 先利用宿主的参考基因组fa序列构建bowtie2索引
bowtie2-build --threads 8 ref.fa ref_bowtie2_index
# 提供宿主参考基因组的bowtie2的索引文件,上一步质控好的双端序列,输出比对后的sam文件
bowtie2 -x ref_bowtie2_index -1 sample_clean.1.fastq.gz -2 sample_clean.2.fastq.gz -S sample.sam 2>sample.mapping.log
# 提取未比对到宿主的序列, -f 4表示:过滤掉未比对到参考基因组的reads(Flag=4)
samtools fastq -@ 8 -f 4 sample.sam -1 sample.unmap.1.fastq.gz -2 sample.unmap.2.fastq.gz -s sample.unmap.single.fastq.gz
步骤3:样本微生物群落组成分析
物种组成分析的方法很多,比如有直接基于reads的分析流程,基于序列组装-基因预测-物种注释的分析流程,以及binning的分析流程等等。这里我演示利用的软件是metaphlan,是基于reads的分析方法。
# metaphlan 运行物种注释
zcat sample.unmap.1.fastq.gz sample.unmap.2.fastq.gz|metaphlan --input_type fastq --bowtie2out sample_bowtie2.bz2 --output_file sample_metaphlan.tsv --nproc 8
# 获取不同物种分类上的结果操作,merge_metaphlan_tables.py 程序是 metaphlan 自带的程序
merge_metaphlan_tables.py *.tsv > 00_merged_abundance_table.txt
grep -E '(p__)|(clade_name)' 00_merged_abundance_table.txt |grep -v 'c__'|sed 's/|/;/g' > 01_metaphlan_phylum.txt
grep -E '(c__)|(clade_name)' 00_merged_abundance_table.txt |grep -v 'o__'|sed 's/|/;/g' > 02_metaphlan_class.txt
grep -E '(o__)|(clade_name)' 00_merged_abundance_table.txt |grep -v 'f__'|sed 's/|/;/g' > 03_metaphlan_order.txt
grep -E '(f__)|(clade_name)' 00_merged_abundance_table.txt |grep -v 'g__'|sed 's/|/;/g' > 04_metaphlan_family.txt
grep -E '(g__)|(clade_name)' 00_merged_abundance_table.txt |grep -v 's__'|sed 's/|/;/g' > 05_metaphlan_genus.txt
grep -E '(s__)|(clade_name)' 00_merged_abundance_table.txt |grep -v 't__'|sed 's/|/;/g' > 06_metaphlan_species.txt
步骤4:序列组装
序列组装的目的是将测序得到的短序列拼接成更长的序列,从而可以在后续分析中获得更完整的信息。这里,对步骤2中去除宿主后的序列,利用megahit软件进行组装,并过滤掉了序列长度小于500bp的组装序列。
# megahit 运行序列组装
megahit -1 sample.unmap.1.fastq.gz -2 sample.unmap.2.fastq.gz -o sample_megahit --out-prefix sample -t 8
# seqkit 过滤小于500bp长度的组装序列
seqkit seq -m 500 sample_megahit/sample.contigs.fa --remove-gaps > sample.contigs_500.fa
sed -i 's/>/>sample_/g' sample.contigs_500.fa
步骤5:基因预测
这一步,我们利用prodigal这一用于原核生物基因预测的软件,可以从上述组装序列中识别和预测基因。
# prodigal 运行基因预测,需要注意-p参数,表示更适用于单菌基因组数据还是meta数据
prodigal -p meta -a sample_prot.faa -m -d sample_nucl.fna -o sample_genes.gff -f gff -s sample.stat -i sample.contigs_500.fa
步骤6:去除冗余基因并生成非冗余基因集
宏基因组项目一般是多个样本进行分析,我们在前面的分析步骤中,都是以单个样本分析作为示例说明,到这一步骤前你会分析得到多个样本的基因预测结果。但是在基因预测完成之后,不同的样本中可能会存在冗余相同的基因,所以我们先把多个样本预测得到的基因序列文件合并,并用cd-hit构建非冗余基因集。
# 合并多个样本的基因序列(蛋白、DNA)
cat sample1_prot.faa sample2_prot.faa ... > prot.faa
cat sample1_nucl.fna sample2_nucl.fna ... > nucl.fna
# cd-hit 运行构建非冗余基因集(蛋白)
cd-hit -i prot.faa -o prot_nonerude.faa -c 0.95 -T 8 -n 5 -d 0 -aS 0.9 -g 1 -sc 1 -sf 1 -M 0
# 获取了非冗余基因名称列表,再通过seqtk把对应的非冗余基因集(DNA)序列挑选出来
grep '>' prot_nonerude.faa|awk -F ' ' '{print $1}'|sed 's/>//g' > prot_nonerude.list
seqtk subseq nucl.fna prot_nonerude.list > nucl_nonerude.fna
# 构建非冗余基因集的bwa索引
bwa index nucl_nonerude.fna -p geneset_bwa
bioawk -c fastx '{print $name, length($seq)}' nucl_nonerude.fna > geneset_length.txt
步骤7:功能注释
对非冗余基因集进行具体的功能注释,可以利用emapper可以得到每个基因的具体功能注释结果。这里主要展现KEGG数据库中的代谢通路和基因功能信息。emapper的输入数据类型有4种:proteins蛋白序列、CDS序列、genome基因组序列、metagenome宏基因组序列。具体来讲,在不利用其基因预测的功能的前提下(软件本身嵌套的也是prodigal),直接输入蛋白序列,emapper共有3个步骤来做功能注释。
Search:序列比对。软件提供了3种模式,分别是:利用Diamond进行序列比对、利用HMMER隐马尔可夫模型进行目标序列检索、利用MMseqs2进行序列比对。可以利用参数-m进行选择。经过search,每条输入的序列会有对应的匹配结果,即seed orthologs。
Orthology inference:直系同源关系推断。上一步序列比对的匹配序列,会对应eggNOG数据库中的蛋白(OG),首先进行大的物种分类学的过滤,然后再根据数据库提供的进化树信息提取更加精细的直系同源关系基因。
Annotation:功能注释。同源关系的推断,在后续可以用于基因功能的注释,可以将已知功能基因的注释信息给未知功能的基因,也就是利用同源关系进行功能注释。也就可以确定我们样本中基因的功能注释结果,这里提供了KEGG、GO、COG等常见功能数据库的注释结果。
利用分析结果中的eggnog.emapper.annotations文件来获取基因功能注释结果。
# emapper 运行基因功能注释
emapper.py -i prot_nonerude.faa -o eggnog --cpu 0 --usemem
# 利用emapper输出结果文件,可以获取功能基因和代谢通路的结果
cut -f1,12 eggnog.emapper.annotations|grep -v "^#"|sed 's/ko://g'|sed '1i gene\tko'|grep -v "-" > KEGG_KO.txt
cut -f1,13 eggnog.emapper.annotations|grep -v "^#"|sed '1i gene\tpathway'|grep -v "-" > KEGG_PATHWAY.txt
步骤8:微生物功能组成分析
在上述步骤,我们得到的是基因的种类,以及具体每个基因的功能注释结果。那么这些基因在每个样本中的具体含量组成是多少呢?只需要将去除宿主序列后(步骤2)的序列与非冗余基因集比对即可,这样我们就可以得到每个样本中,每个基因的含量,也就是样本的功能组成结果表。
# 利用bwa将测序数据和非冗余基因集进行比对,利用samtools获取基因数目
bwa mem -t 4 geneset_bwa sample.unmap.1.fastq.gz sample.unmap.2.fastq.gz | samtools view -bS - | samtools sort - > sample_mapping_geneset.bam
samtools view -F 4 -F 256 -F 2048 sample_mapping_geneset.bam|awk '{if($3!="*") print $3}'|sort| uniq -c|awk 'BEGIN {FS=" ";OFS=","} {print $2,$1}' | awk 'BEGIN {FS=",";OFS=","} {if ($2 > 1) print $1"\t"$2; else print $1"\t0"}'|sed '1i gene\tsample' > sample.count
步骤9:功能结果整理
对步骤7得到的功能结果进一步进行整理,这里我利用了自己写的py程序来做这件事,具体可以见我文章中提供的github仓库,当然大家也可以用其他方法整理成自己想要的结果。
# 这里是我写的py程序的帮助文档,具体用到的各种数据都可以在github仓库中找到
$python kegg.py -h
usage: python kegg.py -kk KEGG_KO.txt -kp KEGG_PATHWAY.txt -mt merged_file.txt -ok out_KO.xls -op out_pathway.xls
Merge KO/pathway count table from eggnog result.
optional arguments:
-h, --help show this help message and exit
-kk KEGGKO, --kegg_KO KEGGKO
Sample's kegg KO information, such as KEGG_KO.txt
-kp KEGGPATHWAY, --kegg_pathway KEGGPATHWAY
Sample's kegg pathway information, such as
KEGG_PATHWAY.txt
-mt MERGETABLE, --merge_table MERGETABLE
Sample's merged gene count table, such as
merged_file.txt
-ok OUTKO, --out_KO OUTKO
Output KO result, such as out_KO.xls
-op OUTPATHWAY, --out_pathway OUTPATHWAY
Output pathway result, such as out_pathway.xls
-t TMP, --tmp TMP Tmp files dir
输出结果的结构
在文件夹00-result文件夹中可以找到主要的输出结果,即本教程说明的终点:样本物种及功能组成表。
├── 00-result # most important results in this fold
│ ├── 00_merged_abundance_table.txt # composition of microbial communities
│ ├── 01_metaphlan_phylum.txt # communities in phylum level
│ ├── 02_metaphlan_class.txt # communities in class level
│ ├── 03_metaphlan_order.txt # communities in order level
│ ├── 04_metaphlan_family.txt # communities in family level
│ ├── 05_metaphlan_genus.txt # communities in genus level
│ ├── 06_metaphlan_species.txt # communities in species level
│ ├── KO_samples.xls # KEGG KO gene composition table
│ └── pathway_samples.xls # KEGG pathway composition table
├── 01-fastp_trim
├── 02-ref_remove
├── 03-metaphlan
├── 04-megahit
├── 05-prodigal
├── 06-cdhit
├── 07-emapper
├── 08-sam_count
├── 09-emapper_kegg
文档信息
- 本文作者:Wanjin Hu (胡万金)
- 本文链接:https://wanjinhu.github.io/2024/02/18/article-metagenomic/
- 版权声明:自由转载-非商用-非衍生-保持署名(创意共享3.0许可证)